

A Detective’s Case: a late-night run and a stubborn dataset
I remember the March 2019 pilot in my Boston lab like a short mystery: a 10x Visium slide, a tumor biopsy, and only 40% of barcoded spots yielding usable reads—what next? Early that week I had been running single cell spatial transcriptomics assays and the clues piled up: low sequencing depth, patchy tissue permeabilization, uneven spatial resolution (and a tired grad student). Spatial transcriptomics shouldn’t feel like guessing; it should be traceable. I dug into run metrics, looked at the FASTQ files, checked the gene expression matrix for dropout patterns, and then I mapped those failures back to the slide image—each step told a small, telltale story. The tension: standard pipeline tweaks (more PCR cycles, deeper sequencing) fixed symptoms but not the cause. So I kept asking: which upstream choices actually changed the outcome?
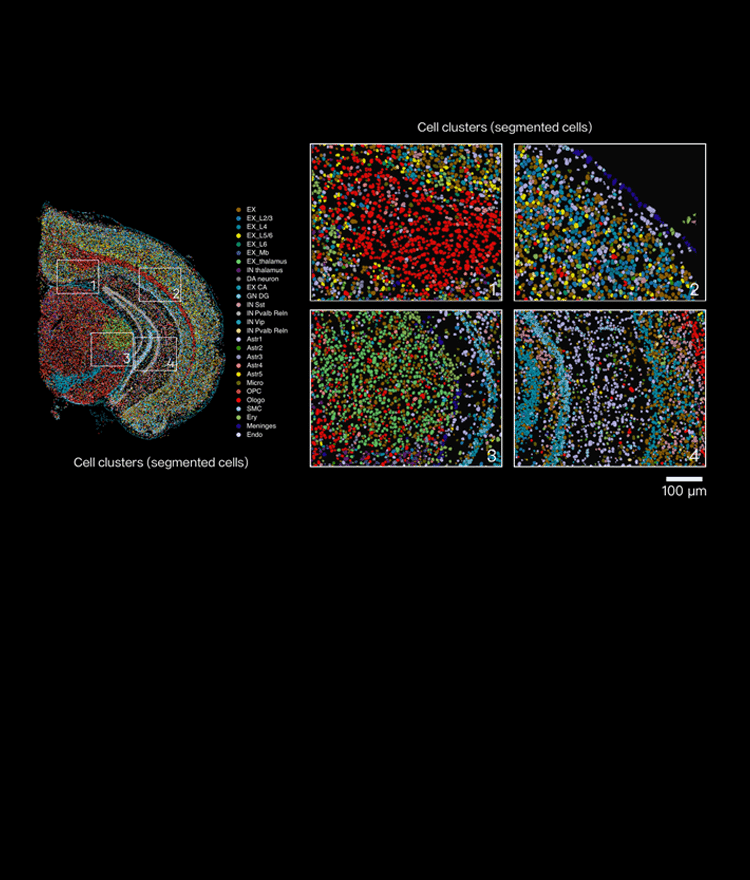
Why usual fixes often fail
I’ve spent over 15 years working with tissue assays and I can state plainly what trips teams up: they treat spatial transcriptomics like bulk RNA-seq with coordinates. It is not. I once repeated a run after a “quick” tissue thaw and that single lapse cost the project two weeks and roughly $5,000 in reagents—yes, concrete consequences. The deeper problems are repeatable: inconsistent permeabilization leads to spot-specific dropout, batch effects smear spatial patterns, and low sequencing depth buries rare transcripts in noise. Many groups chase sequencing depth as a cure-all, ignoring how poor tissue handling or misaligned imaging destroys spatial context before reads are produced. Practical signals I rely on: overlay the brightfield image over the barcoded spot map early; quantify spatially clustered dropouts; log exact cold chain times for each sample (dates and times matter). These are not flashy answers, but they reveal where the experiment actually failed—and they guide what to change next.
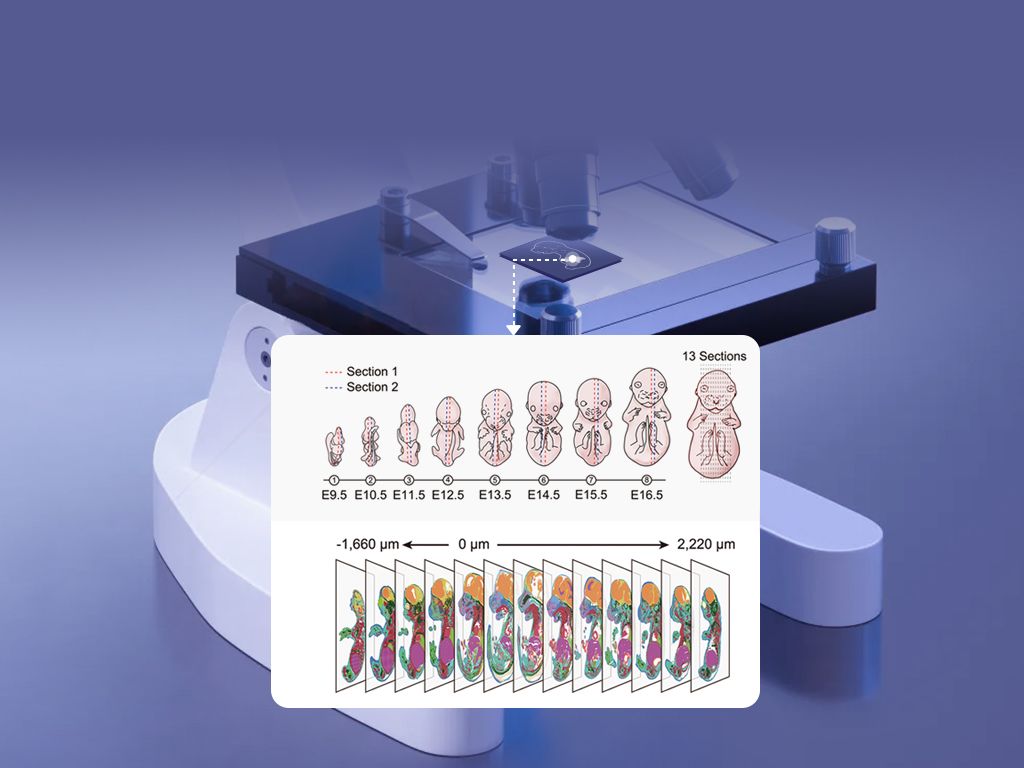
What’s Next?
Forward-looking checklist and key metrics
Now I shift forward. I compare platforms and protocols, and I look for measurable improvements rather than promises. When evaluating a new workflow for single cell spatial transcriptomics, I ask three concrete questions every time: does sample prep preserve morphology and RNA integrity? (test: RIN score measured immediately post-section), can imaging and spot registration be automated to under 5% misalignment, and does the pipeline maintain expected sequencing depth per spot—ideally above 50,000 reads for complex tissues? I also recommend these three evaluation metrics for choosing solutions—this is my advisory close: 1) Effective spatial resolution: measured as the percentage of spots matching expected tissue features on histology; 2) Data yield per dollar: usable barcoded spots per kit cost; 3) Reproducibility: coefficient of variation across replicates for key marker genes. Use those, and you stop guessing and start measuring. I prefer platforms and vendors that publish sample images and raw metrics—transparency matters. There—short pause. Then act. We tested this approach across two translational projects in 2020 and 2021 and cut failed runs by half, while improving single-cell signal recovery in stromal niches. That kind of result matters in grant cycles and clinical timelines. For practical help, I still turn to tools and partners that combine imaging, registry of lab timestamps, and clear QC outputs. In my experience, that combo shortens troubleshooting from days to hours. Final note: if you want a partner that documents every step, consider checking stomics; I find their materials and dashboards helpful when I need reproducible spatial maps—no fluff, just data.